A aorta é a maior artéria do organismo, contribuindo substancialmente para a circulação sistêmica[1]. Anatomicamente, ela pode ser dividida em: raiz da aorta, aorta torácica ascendente, arco aórtico, aorta torácica descendente e aorta abdominal. Do ponto de vista histológico, ela se subdivide em três camadas: a túnica íntima, composta por endotélio; a túnica média, formada por células musculares, fibras elásticas e colágeno; e a túnica adventícia, constituída por tecido conjuntivo, fibroblastos, vasos e nervos. As principais doenças da aorta incluem os aneurismas e as dissecções desse vaso e apresentam uma prevalência global entre 1 e 3%, com incidência crescente conforme o envelhecimento[2].
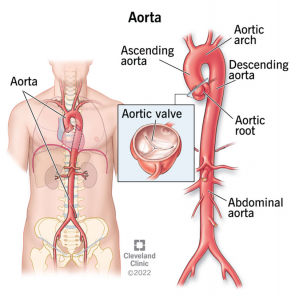
Fonte: Cleveland Clinic, 2022[3].
Classicamente, aneurismas são caracterizados por uma dilatação maior que 1,5 vez o diâmetro do vaso. Nos aneurismas de aorta, eles são representados por um diâmetro axial > 5 cm para a aorta ascendente e > 4 cm para a porção descendente desse vaso[4]. Por sua vez, a dissecção de aorta corresponde à síndrome aórtica aguda mais comum, de elevadas morbidade e mortalidade, caracterizando-se pela criação de uma falsa luz no interior da aorta, uma divulsão entre a camada íntima e a camada média desse vaso causada por uma falha de continuidade na túnica íntima[1]. A mortalidade associada à dissecção de aorta é elevada, estimando-se que entre 1 e 2% dos pacientes com dissecção de aorta ascendente faleçam a cada hora após o início dos sintomas. Assim, o diagnóstico e a intervenção cirúrgica precoce são determinantes para a sobrevida desses pacientes[1].
A relevância dessa emergência médica é tamanha que o dia 19 de setembro foi definido como o Dia Mundial de Conscientização sobre Doenças da Aorta. Atualmente existem diversas iniciativas a fim de chamar a atenção para a identificação precoce dessa doença por meio do reconhecimento de sintomas sugestivos, como o projeto Think Aorta, idealizado pela Society for Cardiothoracic Surgery e o Royal College of Emergency Medicine, nacionalmente conhecida como Pense Aorta[5].

Fonte: Heart Research UK[5]
Atualmente, a medicina vive avanços jamais vistos na área de pesquisa, destacando-se a medicina de precisão. Dentro da medicina de precisão, informações genéticas podem ser usadas para um cuidado mais preciso, aliando-se informações clínicas, histórico familiar e dados genéticos[6]. Nesse contexto, uma grande variedade de variantes patogênicas e/ou provavelmente patogênicas em mais de 30 genes se associam com a fisiopatologia do aneurisma e da dissecção de aorta; assim, o teste genético, quando bem indicado, pode auxiliar no diagnóstico precoce de quadros assintomáticos, trazendo ganhos à qualidade de vida desses pacientes[7].
Os principais genes associados às doenças hereditárias da aorta são codificadores de proteínas constituintes da matriz extracelular, da via de sinalização do fator de transformação do crescimento beta (TGF-ß) e das células musculares lisas[2]. O principal painel genético para investigação de doenças hereditárias da aorta torácica inclui 11 genes prioritários, por apresentarem alto risco de penetrância: FBN1, LOX, COL3A1, TGFBR1, TGFBR2, SMAD3, TGFB2, ACTA2, MYH11, MYLK e PRKG1[8].

Do ponto de cista clínico, as doenças hereditárias da aorta torácica são classificadas nas formas sindrômicas, representando 5% dos casos, e não sindrômicas, com uma prevalência de 95%[1]. As formas sindrômicas se definem pelo acometimento de outros sistemas orgânicos além do cardiovascular, enquanto as formas não sindrômicas são restritas à artéria aorta[9]. Dentre as formas sindrômicas, destacam-se a síndrome de Marfan, a síndrome de Loeys-Dietz e a síndrome de Ehlers-Danlos[1].
Síndrome de Marfan
A síndrome de Marfan é o fenótipo de doenças hereditárias da doença aorta mais prevalente, acometendo aproximadamente 1 em cada 5 mil para 1 em cada 10 mil indivíduos. Apresenta herança autossômica dominante associada a mutações no gene fibrilina-1 (FBN1) e acometimento sistêmico, com manifestações cardiovasculares, esqueléticas, oftalmológicas, neurológicas, dermatológicas e pulmonares[10].
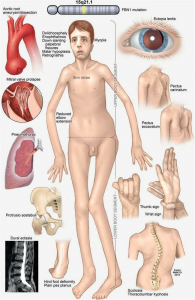
Síndrome de Loeys-Dietz
A síndrome de Loeys-Dietz é uma fenocópia da síndrome de Marfan e da síndrome de Ehlers-Danlos. Apresenta herança autossômica dominante associada a mutações em genes codificadores do fator de transformação do crescimento beta 1 e 2 (TGFBR1 e TGFBR2) e dos genes SMAD3 e TGFB2[12]. O diagnóstico clínico desses pacientes envolve a identificação de aneurismas arteriais, hipertelorismo, úvula bífida ou fenda palatina[13].
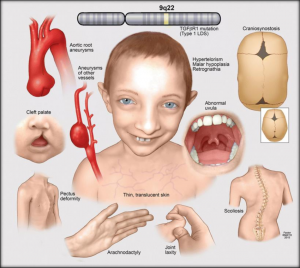
Fonte: Suleiman, 2023[11].
A síndrome vascular de Ehlers-Danlos é uma desordem do tecido conjuntivo caracterizada por desequilíbrios na formação das moléculas de colágeno, com manifestações sistêmicas. O fenótipo vascular dessa síndrome representa uma condição autossômica dominante majoritariamente associada a mutações no gene COL3A1[14].
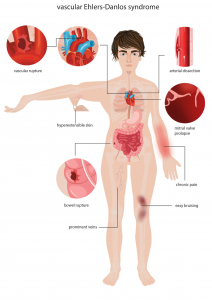
As formas não sindrômicas podem ser subdivididas na forma familiar (quando atingem 2 ou mais familiares na mesma família) ou esporádica[16]. Nesses pacientes, as formas familiares dos aneurismas da aorta torácica estão associadas principalmente a variantes genéticas nos genes ACTA2, MYH11, PRKG1 e MYLK[1,2].
Existem ainda formas congênitas inseridas na fisiopatologia dos aneurismas e dissecções, em especial da aorta ascendente e torácica, como a válvula aórtica bicúspide e a síndrome de Turner[1].
Válvula aórtica bicúspide
Casos de válvula aórtica bicúspide são mais incidentes no sexo masculino, apresentando caráter esporádico ou uma herança autossômica dominante. Nesses pacientes, faz-se necessário o acompanhamento regular com exames de imagem, de periodicidade variável ao diâmetro da aorta, por sua associação com dilatação e dissecção da aorta, especialmente em idades mais avançadas, pelo risco de crescimento tardio desse vaso[1].

Fonte: Rashed et al., 2022[17].
Síndrome de Turner
A deleção parcial ou total de um dos pares do cromossomo X é causadora da síndrome de Turner em pacientes do sexo feminino, com prevalência de 1 a cada 2.500 nascidas vivas[18]. Dilatação e coartação de aorta, hipertensão arterial e válvula aórtica bicúspide são fatores predisponentes de dissecção da aorta nessas pacientes, em geral ocorrendo a diâmetros menores, pela baixa estatura associada a essa condição[1].
A mais recente diretriz da European Society of Cardiology (2024), juntamente com a diretriz do American College of Cardiology/American Heart Association (2022), traz atualizações acerca das indicações de teste genético em pacientes com aneurisma ou dissecção de aorta, bem como do rastreamento familiar das variantes de risco identificadas nos casos-índice. Assim, o teste genético é recomendado para pacientes com aneurismas da raiz da aorta e/ou de sua porção ascendente ou dissecção da aorta torácica que apresentem fatores de risco para doenças hereditárias da aorta torácica, como manifestações sindrômicas, histórico familiar positivo e início da sintomatologia em idades jovens. Diante de um resultado positivo do teste genético, com identificação de variantes patogênicas ou provavelmente patogênicas, deve-se fazer o rastreamento em cascata dos familiares. Todo o processo deve ser permeado por um aconselhamento genético pré e pós-teste, com uma equipe multiprofissional especializada. Para indivíduos com tais aortopatias, porém sem identificação de uma variante causal por meio do teste genético, o screening familiar deve seguir com exames de imagem, destacando-se o papel do ecocardiograma transtorácico[1,2].
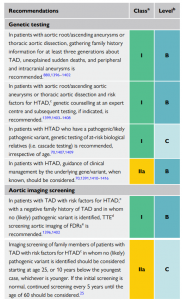
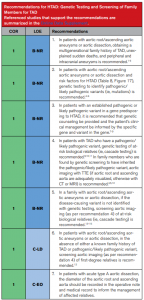
Assim, conclui-se que as doenças da aorta apresentam fenótipo diverso e genótipo variado, sendo o estudo genético desses pacientes uma ferramenta de valor prognóstico e aplicação crescente na prática médica atual.
Referências
- Isselbacher EM, Preventza O, Hamilton Black J 3rd, Augoustides JG, Beck AW, Bolen MA, et al.; Peer Review Committee Members. 2022 ACC/AHA Guideline for the Diagnosis and Management of Aortic Disease: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. Circulation. 2022;146:e334-e482. doi: 10.1161/CIR.0000000000001106.
- Mazzolai L, Teixido-Tura G, Lanzi S, Boc V, Bossone E, Brodmann M, et al.; SC Scientific Document Group. 2024 ESC Guidelines for the management of peripheral arterial and aortic diseases. Eur Heart J. 2024;1-163. doi: 10.1093/eurheartj/ehae179.
- Cleveland Clinic. Aorta. 2022. Disponível em: https://my.clevelandclinic.org/health/body/17058-aorta-anatomy.
- Munden RF, Carter BW, Chiles C, MacMahon H, Black WC, Ko JP, et al. Managing incidental findings on thoracic CT: mediastinal and cardiovascular findings. A white paper of the ACR Incidental Findings Committee. J Am Coll Radiol. 2018;15(8):1087-96.
- Heart Research UK. Think Aorta campaign update. Disponível em: https://heartresearch.org.uk/think-aorta/.
- Wang RS, Maron BA, Loscalzo J. Multiomics network medicine approaches to precision medicine and therapeutics in cardiovascular diseases. Arterioscler Thromb Vasc Biol. 2023;43(4):493-503. Disponível em: <https://pubmed.ncbi.nlm.nih.gov/36794589/>.
- Ashvetiya T, Fan SX, Chen YJ, Williams CH, O’Connell JR, Perry JA, Hong CC. Identification of novel genetic susceptibility loci for thoracic and abdominal aortic aneurysms via genome-wide association study using the UK Biobank Cohort. PLoS One. 2021;16(9):e0247287.
- Renard M, Francis C, Ghosh R, Scott AF, Witmer PD, Adès LC, et al. Clinical validity of genes for heritable thoracic aortic aneurysm and dissection. J Am Coll Cardiol. 2018;72;605-15.
- Faggion Vinholo T, Zafar MA, Ziganshin BA, Elefteriades JA. Nonsyndromic thoracic aortic aneurysms and dissections-is screening possible? Semin Thorac Cardiovasc Surg. 2019;31(4):628-34. Disponível em: https://www.sciencedirect.com/science/article/pii/S1043067919300826?via%3Dihub.
- Hernándiz A, Zúñiga A, Valera F, Domingo D, Ontoria-Oviedo I, Marí JF, Román JA, et al. Relação genótipo FBN1/fenótipo em uma coorte de pacientes com síndrome de Marfan. Clin Genet. 2021;99(2):269-80.
- Suleiman M. The Use of Bioelectrical Impedance Analysis as a Contemporary Biomarker in Congenital or Hereditary Diseases with Aortic Involvement. 2023 [tese de doutorado]. Disponível em: https://www.researchgate.net/publication/367189309_The_Use_of_Bioelectrical_Impedance_Analysis_as_a_Contemporary_Biomarker_in_Congenital_or_Hereditary_Diseases_with_Aortic_Involvement?_tp=eyJjb250ZXh0Ijp7ImZpcnN0UGFnZSI6Il9kaXJlY3QiLCJwYWdlIjoiX2RpcmVjdCJ9fQ.
- Malyuk DF, Campeau N, Benson JC. Loeys-Dietz syndrome: Case report and review of the literature. Radiol Case Rep. 2022;17(3):767-70.
- MacCarrick G, Black JH 3rd, Bowdin S, El-Hamamsy I, Frischmeyer-Guerrerio PA, Guerrerio AL, et al. Loeys-Dietz syndrome: a primer for diagnosis and management. Genet Med. 2014;16(8):576-87.
- Wei X, Zhou X, Xie B, Shi M, Gui C, Liu B, et al. Importance of comprehensive genetic testing for patients with suspected vascular Ehlers-Danlos syndrome: a family case report and literature review. Front Genet. 2023;14:1246712.
- Progenics. Ehlers-Danlos. Disponível em: https://www.progenics.com.au/diseases/ehlers-danlos.
- Rohde S, Zafar MA, Ziganshin BA, Elefteriades JA. Thoracic aortic aneurysm gene dictionary. Asian Cardiovasc Thorac Ann. 2021;29(7):682-96.
- Rashed ER, Dembar A, Riasat M, Zaidi AN. Bicuspid aortic valves: an up-to-date review on genetics, natural history, and management. Curr Cardiol Rep. 2022;24(8):1021-30. doi: 10.1007/s11886-022-01716-2.
- Shankar Kikkeri N, Nagalli S. Turner Syndrome. [Updated 2023 Aug 8]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Disponível em: https://www.ncbi.nlm.nih.gov/books/NBK554621/.