As doenças congênitas da aorta podem ser divididas em 2 categorias principais: aquelas de apresentação precoce, no período neonatal, e aquelas com manifestações tardias, na infância e na vida adulta. As lesões de manifestações precoces são geralmente de natureza obstrutiva e incluem coarctação da aorta, síndrome do coração esquerdo hipoplásico e anéis vasculares, tendendo a envolver o arco da aorta. Os problemas congênitos da aorta de início tardio são principalmente aneurismáticos e envolvem predominantemente a aorta ascendente, além da valva aórtica bicúspide congênita.
A correção cirúrgica de outras cardiopatias pode apresentar complicações aórticas, geralmente de natureza aneurismática. Esses são problemas particularmente desafiadores devido à escassez de dados nos quais basear as decisões. Esses aneurismas via de regra seguem após cirurgia para lesões conotruncais, como tetralogia de Fallot (TF), truncus arteriosus communis persistente e ventrículo direito de saída dupla (VDSD), mas também após o procedimento de Norwood, a cirurgia de Jatene e o procedimento de Ross.
COARTAÇÃO DA AORTA
A maioria das técnicas atuais para reparo de coarctação da aorta e interrupção completa do arco aórtico – obstruções que ocorrem mais frequentemente apenas distalmente à artéria subclávia esquerda – têm sido usadas nos últimos 30 anos e geralmente envolvem a ressecção completa do segmento envolvido seguido de anastomose primária da aorta proximal e distal. O reparo primário permite que a aorta cresça de forma adequada e paralela ao crescimento somático. Se ocorrer uma obstrução subsequente, muitas vezes isso pode ser tratado com sucesso por via minimamente invasiva/transcateter.
Algumas das abordagens anteriores à obstrução do arco aórtico envolviam aumento do segmento estreito ou enxertos de interposição usando materiais protéticos como o Dacron, frequentemente com ressecção incompleta do segmento estenótico e seu tecido doente. Reparos envolvendo Dacron não resultaram apenas em obstrução residual ocasional, mas também em incidência alarmante de aneurisma tardio e/ou dissecção da aorta. Em estudo recente de centro único da Universidade de Wisconsin, 10% dos pacientes com correção de patch apresentaram aneurismas em 10 anos, e mais de 50% apresentaram em 25 anos; 14% destes sofreram ruptura dentro de 20 anos, e metade desses pacientes morreu. Isso sugere que a vigilância rigorosa dos pacientes com aortoplastia com patch de Dacron é obrigatória e a reintervenção precoce é justificada.
DILATAÇÃO AÓRTICA APÓS REPARO DE LESÕES CONOTRUNCAIS
Com a melhora nos resultados das correções cirúrgicas das cardiopatias congênitas, há agora mais adultos vivendo com doença cardíaca congênita do que crianças. Entre esses, estão os pacientes que foram submetidos a reparo de defeitos conotruncais (isto é, TF, VSDS e truncus arteriosus), ao procedimento de cirurgia de Jatene para a transposição completa das grandes artérias (TGA), ao procedimento de Norwood para a síndrome do coração esquerdo hipoplásico (SHCE) e ao procedimento de Ross para doença da válvula aórtica. Em todas essas condições, a aorta tende a dilatar, apresentando risco teórico de ruptura e/ou dissecção tardia, maior probabilidade de incompetência da válvula aórtica e possibilidade de compressão de estruturas adjacentes. O tamanho da aorta em alguns pacientes com o procedimento de Fontan, no qual o fluxo sanguíneo pulmonar é fornecido passivamente pelo sistema venoso sistêmico, apresenta problema particular, pois a aorta dilatada pode comprimir as artérias pulmonares e obstruir o retorno venoso sistêmico.
De uma perspectiva embriológica, podemos ver todas essas anomalias congênitas como decorrentes de várias perturbações da partição das vias de saída do coração. A via de saída é inicialmente um tubo único, com o cone do ventrículo direito levando ao truncus arteriosus. Essa via de saída é normalmente dividida pelos septos aorticopulmonares e conotruncais combinados em canais aproximadamente iguais, fornecendo fluxo pulmonar do ventrículo direito e fluxo aórtico do ventrículo esquerdo.
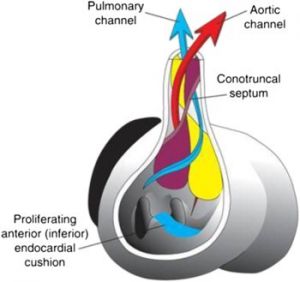 (FIG 1. Desenvolvimento cardíaco normal)
(FIG 1. Desenvolvimento cardíaco normal)
Talvez o exemplo mais facilmente compreendido de divisão desequilibrada do conotruncus seja a TF. Nessa condição, o septo entre a saída do ventrículo direito para a artéria pulmonar e a saída do ventrículo esquerdo para a aorta é desviado muito para a direita, levando à obstrução da via de saída do ventrículo direito e resultando em vários graus de estenose ou atresia pulmonar. O mau posicionamento do septo também resulta em uma aorta grande e evita que a porção infundibular do septo conotruncal/aortopulmonar se conecte com o septo interventricular muscular/membranoso inferiormente, levando ao defeito do septo ventricular de mau alinhamento característico.
Se o septo conotruncal/aortopulmonar for deslocado muito para a esquerda, ele restringe severamente o fluxo aórtico, com efeitos devastadores no desenvolvimento do ventrículo esquerdo, aorta ascendente e arco aórtico, levando a SHCE, interrupção do arco aórtico e várias formas de coarctação. Outro tipo de anormalidade conotruncal é a TGA, na qual o septo não consegue realizar sua rotação espiral usual durante o desenvolvimento, resultando em um ventrículo direito que deságua na aorta e um ventrículo esquerdo com saída para a artéria pulmonar. A TGA completa é uniformemente fatal sem intervenção, a menos que uma lesão mista esteja presente, como um canal arterial patente ou um defeito do septo atrial ou ventricular.
Independentemente da natureza da anormalidade da septação, os indivíduos com essas lesões conotruncais apresentam aorta anormalmente menos resistente e frequentemente aumentada desde a infância.
O exame histológico revela que há alguma fragmentação e perda das lamelas elásticas presentes já ao nascimento, que também progride com a idade. O grau de ruptura da arquitetura normal da parede aórtica se correlaciona aproximadamente com o grau de dilatação aórtica.
A maioria dos dados disponíveis vem de pacientes com as lesões conotruncais mais comuns, principalmente TF, porque esses pacientes historicamente tiveram os primeiros reparos e viveram mais. Todos os pacientes com TF têm aorta dilatada ao nascimento e, independentemente da idade na cirurgia e do tipo de intervenção, a aorta continua a aumentar ao longo da vida. Embora essas aortas permaneçam grandes mesmo após o reparo intracardíaco precoce e continuem a crescer ao longo da vida, há apenas raros casos em que ruptura ou dissecção foram relatados. Nos casos relatados de reparo cirúrgico, a substituição da aorta foi geralmente necessária mais de 20 anos após o reparo inicial da TF e geralmente por incompetência aórtica moderada ou grave, em vez de ruptura ou dissecção. Idade avançada no reparo inicial, sexo masculino e atresia pulmonar parecem ser fatores de risco para aneurisma da aorta em pacientes com TF, mas há poucos casos para gerar diretrizes confiáveis para intervenção eletiva para aorta dilatada na TF.
Dilatação da aorta após os procedimentos de cirurgia de Jatene, Norwood e Ross
A cirurgia de Jatene – uma das maiores histórias de sucesso no tratamento de doenças cardíacas congênitas – proporcionou expectativa e qualidade de vida quase normais a pacientes com TGA completa, resgatando-os da história natural sombria e uniformemente fatal. A operação de troca arterial é tecnicamente exigente, porém apresenta um alto nível de sucesso, tanto no início quanto no final do reparo.
Embora seja frequentemente chamada de correção anatômica, na verdade não é: as grandes artérias são trocadas de modo que a aorta seja conectada ao ventrículo esquerdo e a artéria pulmonar ao ventrículo direito e as artérias coronárias sejam transferidas, mas as raízes aórtica e pulmonar (incluindo suas respectivas válvulas semilunares), permanecem não comutadas. Além das diferenças intrínsecas entre as 2 válvulas semilunares e o tecido sinusal que as circunda, a raiz de cada grande artéria é distorcida pela remoção das artérias coronárias do remendo dos defeitos (no caso da aorta), e o reimplante dessas artérias na raiz pulmonar (neo-aórtica). A dilatação da raiz neo-aórtica/aorta ascendente reconstruída é comum após o procedimento de cirurgia de Jatene, contudo a insuficiência clinicamente importante da válvula pulmonar – na posição aórtica – é surpreendentemente rara: ocorre em menos de 5%. As imagens dos aneurismas da aorta ascendente em pacientes com cirurgia de Jatene costumam ser bastante impressionantes, mas nenhum caso de ruptura ou dissecção foi relatado. Dados do Children’s Hospital of Boston mostram que dilatação acentuada da raiz aórtica está presente em 95% dos pacientes de 17 a 18 anos de idade, entretanto a maioria das válvulas neo-aórticas são competentes, mesmo na presença de graves dilatação da raiz.
A aorta também tende a se tornar aneurismática após o procedimento de Norwood para reparo de SHCE, uma das doenças cardíacas congênitas mais graves, em que as estruturas do lado esquerdo do coração falham em se desenvolver no útero.
Na SHCE, a válvula aórtica é pequena; a aorta ascendente pode ter apenas 3 a 4 mm de diâmetro e o arco aórtico é hipoplásico. Com a paliação de Norwood, a neo-aorta é reconstruída usando uma porção da artéria pulmonar proximal, uma faixa de aorta nativa e material protético – geralmente homoenxerto criopreservado – para que desobstrua o fluxo do ventrículo direito, e fluxo pulmonar confiável e controlado é fornecido diretamente através de um pequeno conduto do ventrículo direito para a confluência da artéria pulmonar (um shunt ”Sano”) ou usando um shunt de Blalock-Taussig modificado da artéria subclávia para a artéria pulmonar. A neo-aorta tende a se dilatar com o tempo, muitas vezes atingindo diâmetros bastante grandes. Nessa população, existe preocupação particular com o aneurisma pressionando as artérias pulmonares e causando obstrução ao fluxo pulmonar. A experiência do Hospital Infantil da Filadélfia mostra que virtualmente todas as neo-aortas tornam-se maciçamente dilatadas, com escores Z de 5 ou mais após 5 a 10 anos de acompanhamento. No entanto, não há casos relatados de ruptura ou dissecção dessas aortas, o que deve moderar nosso entusiasmo pela intervenção profilática na ausência de compressão da artéria pulmonar ou incompetência da válvula neo-aórtica.
Há também uma tendência bem conhecida da aorta de dilatar após o procedimento de Ross, tanto a raiz pulmonar transposta do autoenxerto quanto a aorta ascendente distal nativa, principalmente quando a válvula aórtica é bicúspide. A escassez de casos relatados de dissecção radicular de autoenxerto ou ruptura sugere que uma abordagem conservadora é prudente e que os critérios tradicionais para substituição de aorta ascendente dilatada e raízes podem não ser aplicáveis após o procedimento de Ross. Quando a reintervenção é necessária, geralmente é para regurgitação valvar com autoenxerto; como os folhetos do autoenxerto costumam ser normais ou minimamente distorcidos, esses pacientes podem ser bons candidatos para procedimentos de preservação da válvula. O risco de dilatação da aorta parece ser aumentado em pacientes que são operados em uma idade jovem, aqueles com doença aneurismática na primeira operação, aqueles com regurgitação aórtica e valva aórtica bicuspide e, finalmente, aqueles que são submetidos a substituição completa da raiz da aorta com o autoenxerto pulmonar, ao invés da inserção da válvula pulmonar dentro da aorta nativa.
Risco geral de ruptura ou dissecção em pacientes com síndromes genéticas:
Em geral, o risco de ruptura ou dissecção nas primeiras 2 décadas de vida é relativamente baixo: é extremamente raro na infância, exceto nas síndromes de Loeys-Dietz e Turner.
Valva aórtica biscúspide e a síndrome de Marfan geralmente representam problema apenas no final da adolescência e na idade adulta. Embora haja relatos de ruptura e dissecção durante os primeiros 12 anos de vida, essas catástrofes aórticas são extremamente raras.
A síndrome de Loeys-Dietz é uma exceção à generalização de que as aortopatias geralmente não causam catástrofes em crianças. De fato, a ruptura e a dissecção, podem ocorrer em idades muito jovens (6 meses de idade) e em diâmetros aórticos muito pequenos (quase normais). Este grupo de pacientes, portanto, provavelmente merece uma abordagem profilática cirúrgica mais agressiva.
O outro grupo de risco na infância são os pacientes com síndrome de Turner, que apresenta risco aumentado de dissecção. Um estudo recente do National Institutes of Health com mais de 200 pacientes identificou 3 pacientes que tiveram uma dissecção aórtica: todos tinham dilatação da aorta significativamente maior do que o grupo geral como um todo, mas o diâmetro da aorta na dissecção era inferior a 5 cm. Nesse grupo de pacientes, relacionar o tamanho do aneurisma a superfície corporal pode ser importante.
Z SCORES
Uma palavra de cautela é apropriada sobre o uso de escores z no tratamento da aorta dilatada. O escore z descreve o número de desvios padrão da média de uma determinada medida quando comparado com indivíduos normais com a mesma superfície corporal e, portanto, fornece base para comparação com outras crianças de tamanhos corporais diferentes.
Na extremidade final da distribuição – o percentil 95 ao 97 – uma mudança muito pequena na medida da aorta pode desencadear um salto muito grande no escore z. Nestes casos, é importante manter alguma perspectiva e ter em mente a medida real da aorta: a mudança no escore z pode ser consequência de uma diferença que está dentro da faixa de erro da medida aórtica.
Conclusões
Em resumo, há número substancial e crescente de pacientes com doença cardíaca congênita que foram submetidos a cirurgia corretiva e agora apresentam aortas grandes e com dilatação progressiva, cuja história “natural” não é completamente compreendida. As indicações para cirurgia neste grupo de pacientes, portanto, ainda não são baseadas em evidências. Dessa forma, a abordagem desses pacientes deve ser criteriosa e com máxima cautela, de modo que não substituamos toda aorta dilatada, particularmente em pacientes que tiveram múltiplas esternotomias, colaterais aortopulmonares e corações dilatados e com sobrecarga volumétrica e/ou pressórica prévia. Para esses pacientes a cirurgia é uma perspectiva intimidadora. Entretanto, consideração especial deve ser dada aos pacientes com síndrome de Loeys-Dietz por causa de sua aortopatia particularmente agressiva, e àqueles com síndrome de Turner, nos quais sabemos que a superfície corporal é importante. Nas síndromes de Loeys-Dietz e Turner, a ruptura e a dissecção ocorrem em diâmetros aórticos relativamente pequenos, ainda no início da vida.
Bibliografia:
- Zanotti G, Vricella LA, Cameron DEC. Thoracic aortic aneurysm syndromes in children. Semin Thorac Cardiovasc Surg Pediatr Card Surg Ann. 2008; 11: 11-21
- Cramer JW, Ginde S, Bartz PJ, Tweddell JS, Litwin SB, Earing MG. Aortic aneurysms remain a significant source of morbidity and mortality after use of Dacron patch aortoplasty to repair coarctation of the aorta: results from a single center. Pediatr Cardiol. 2013; 34: 296-301
- Warnes CA, Williams RG, Bashore TM, Child JS, Connolly HM, Dearani JA, et al. ACC/AHA 2008 guidelines for the management of adults with congenital heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2008; 52: 143-263.
- Niwa K, Perloff JK, Bhuta SM, Laks H, Drinkwater DC, Child JS, et al. Structural abnormalities of great arterial walls in the congenital heart disease. Light and electron microscopic analyses. Circulation. 2001; 103: 393-400.
- Niwa K. Aortic root dilatation in tetralogy of Fallot long-term after repair-histology of the aorta in tetralogy of Fallot: evidence of intrinsic aortopathy. Int J Cardiol. 2005; 103: 117-119.
- Schwartz ML, Gauvreau K, del Nido P, Mayer JE, Colan SD. Long-term predictors of aortic root dilation and aortic regurgitation after arterial switch operation. Circulation. 2004; 110: 128-132.
- Cohen MS, Marino BS, McElhinney DB, Robbers-Visser D, van der Woerd W, Gaynor JW, et al. Neo-aortic root dilation and valve regurgitation up to 21 years after staged reconstruction for hypoplastic left heart syndrome. J Am Coll Cardiol. 2003; 42: 533-540.
- Cameron DE, Vricella LA. What is the proper place of the Ross procedure in our modern armamentarium? Curr Cardiol Rep. 2007; 9: 93-98.
- MacCarrick G, Black JH, Bowdin S, El-Hamamsy I, Frischmeyer-Guerrerio PA, Guerrerio AL, et al. Loeys-Dietz syndrome: a primer for diagnosis and management. Genet Med. 2014; 16: 576-587.
- Matura LA, Ho VB, Rosing DR, Bondy CA. Aortic dilatation and dissection in Turner syndrome. Circulation. 2007; 116: 1663-1670.